- Аномалия Арнольда-Киари: симптомы и лечение
- Разновидности аномалии Арнольда-Киари
- Симптомы
- Диагностика
- Лечение
- Аномалия Арнольда-Киари
- Описание болезни
- Причины заболевания
- Проявления
- Аномалия Киари
- Причины возникновения аномалии Киари
- Симптомы аномалии Киари
- Лечение аномалии Киари
- Прогноз аномалии Киари
- Аномалия Арнольда Киари 1 типа – что это? Симптомы, последствия, лечение
- Причины возникновения
- Факторы риска
- Типы синдрома
- Основные симптомы
- Другие признаки
- Осложнения
- Консервативное лечение
- Хирургическое вмешательство
- Профилактика
Аномалия Арнольда-Киари: симптомы и лечение
Аномалия Арнольда-Киари – это нарушение строения и расположения мозжечка, ствола мозга относительно черепа и позвоночного канала. Это состояние относится к врожденным порокам развития, хотя не всегда проявляет себя с первых дней жизни. Иногда первые симптомы появляются после 40 лет.
Аномалия Арнольда-Киари может проявляться различными симптомами поражения головного мозга, спинного мозга, нарушения циркуляции спинномозговой жидкости. Точку в диагностике ставит обычно магнитно-резонансная томография. Лечение проводят консервативными и хирургическими методами.
Из этой статьи Вы сможете узнать подробнее о симптомах, диагностике и способах лечения аномалии Арнольда-Киари.
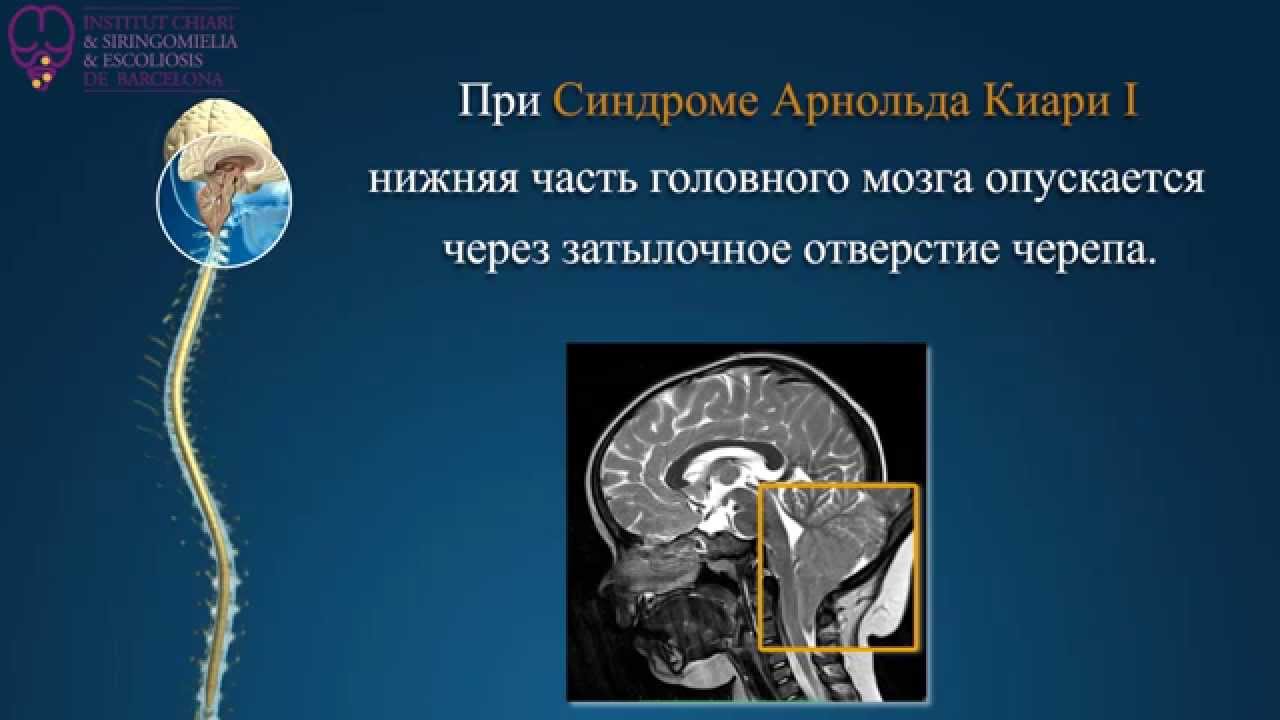
В норме рубеж между головным и спинным мозгом находится на уровне между костями черепа и шейным отделом позвоночника. Здесь располагается большое затылочное отверстие, которое, по сути, и служит условной линией. Условной, потому что ткань головного мозга переходит в спинной мозг не прерываясь, без четкой границы.
Все анатомические структуры, располагающиеся выше большого затылочного отверстия, в частности, продолговатый мозг, мост и мозжечок, относятся к образованиям задней черепной ямки. Если эти образования (по одному или все вместе) спускаются ниже плоскости большого затылочного отверстия, то тогда и возникает аномалия Арнольда-Киари.
Такое неправильное расположение мозжечка, продолговатого мозга приводит к компрессии спинного мозга в области шейного отдела позвоночника, мешает нормальной циркуляции спинномозговой жидкости. Иногда аномалия Арнольда-Киари сочетается с другими пороками развития краниовертебрального перехода, то есть места перехода черепа в позвоночник.
В таких сочетанных случаях симптоматика обычно выражена сильнее и дает о себе знать довольно рано.
Аномалия Арнольда-Киари носит имя двух ученых: австрийского патологоанатома Ханса Киари и немецкого патологоанатома Юлиуса Арнольда. Первый еще в 1891 году описал ряд аномалий развития мозжечка и ствола мозга, второй в 1894 году дал анатомическое описание опущения нижней части полушарий мозжечка в большое затылочное отверстие.
Разновидности аномалии Арнольда-Киари
Согласно статистике, аномалия Арнольда-Киари встречается с частотой от 3,2 до 8,4 случаев на 100 000 населения. Столь широкий диапазон отчасти обусловлен неоднородностью этого порока развития.
О чем идет речь? Дело в том, что аномалию Арнольда-Киари принято делить на четыре подтипа (описаны Киари), в зависимости от того, какие структуры опущены в большое затылочное отверстие и насколько они неправильные по строению:
- аномалия Арнольда-Киари I – когда в позвоночный канал из черепной коробки опускаются миндалины мозжечка (нижняя часть полушарий мозжечка);
- аномалия Арнольда-Киари II – когда опускается в позвоночный канал большая часть мозжечка (в том числе и червь), продолговатый мозг, IV желудочек;
- аномалия Арнольда-Киари III – когда ниже большого затылочного отверстия располагаются почти все образования задней черепной ямки (мозжечок, продолговатый мозг, IV желудочек, мост). Довольно часто они располагаются в мозговой грыже шейно-затылочной области (ситуация, когда есть дефект позвоночного канала в виде незаращения дужек позвонков, и содержимое дурального мешка, то есть спинной мозг со всеми оболочками, выпячивается в этот дефект). Диаметр большого затылочного отверстия в случае такого типа аномалии увеличен;
- аномалия Арнольда-Киари IV – недоразвитие (гипоплазия) мозжечка, но при этом сам мозжечок (вернее то, что образовалось на его месте) располагается правильно.
I и II типы порока встречаются чаще. Это связано с тем, что III и IV типы обычно не совместимы с жизнью, смерть наступает в первые дни жизни.
До 80% всех случаев аномалии Арнольда-Киари сочетается с наличием сирингомиелии (заболевание, характеризующееся наличием в спинном мозге полостей, замещающих мозговую ткань).
В развитии аномалии ведущая роль принадлежит нарушениям формирования структур мозга и позвоночника во внутриутробном периоде.
Однако следует учитывать и следующий фактор: травма головы, полученная в период родов, повторные черепно-мозговые травмы в детском возрасте могут повреждать костные швы в области основания черепа. В результате нормальное формирование задней черепной ямки нарушается.
Она становится слишком маленькой, с уплощенным скатом, из-за чего все структуры задней черепной ямки просто не в состоянии в ней уместиться. Они «ищут выход» и устремляются в большое затылочное отверстие, а далее – в позвоночный канал.
Эта ситуация в какой-то мере считается приобретенной аномалией Арнольда-Киари. Также симптомы, сходные с аномалией Арнольда-Киари, могут возникнуть при развитии опухоли головного мозга, которая заставляет полушария мозжечка сдвинуться в большое затылочное отверстие и позвоночный канал.
Симптомы
Одним из проявлений аномалии Арнольда-Киари может быть гидроцефалия.
Основные клинические проявления аномалии Арнольда-Киари связаны со сдавлением структур мозга. Одновременно сдавливаются сосуды, питающие мозг, пути ликворотока, корешки черепно-мозговых нервов, проходящие в этой области.
Принято выделять 6 неврологических синдромов, которыми может сопровождаться аномалия Арнольда-Киари:
Естественно, что далеко не всегда все 6 синдромов присутствуют. Их выраженность варьирует в той или иной степени, в зависимости от того, какие структуры и насколько сдавливаются.
Гипертензионно-гидроцефальный синдром развивается в результате нарушения циркуляции спинномозговой жидкости (ликвора). В норме ликвор свободно перетекает из субарахноидального пространства головного мозга в субарахноидальное пространство спинного мозга.
Опустившаяся нижняя часть миндалин мозжечка блокирует этот процесс, словно пробка бутылку. Образование ликвора в сосудистых сплетениях головного мозга продолжается, а оттекать, по большому счету, ему некуда (не считая естественных механизмов всасывания, которых в этом случае недостаточно).
Ликвор накапливается в головном мозге, вызывая повышение внутричерепного давления (внутричерепную гипертензию) и расширение ликворосодержащих пространств (гидроцефалию). Это проявляет себя головной болью распирающего характера, которая усиливается при кашле, чихании, смехе, натуживании.
Боль ощущается в затылке, области шеи, возможно напряжение мышц шеи. Могут появляться эпизоды внезапной рвоты, никоим образом не связанной с приемом пищи.
Мозжечковый синдром проявляет себя нарушением согласованности движений, «пьяной» походкой, мимопопаданием при выполнении целенаправленных движений. Больных беспокоит головокружение. Возможно появление дрожания в конечностях.
Может нарушаться речь (становится разделенной на отдельные слоги, скандирующей). Довольно специфическим симптомом считается «нистагм, бьющий вниз». Это непроизвольные подергивания глазных яблок, направленные, в данном случае, книзу.
Больные могут жаловаться на двоение в глазах из-за нистагма.
Бульбарно-пирамидный синдром носит такое название по наименованию структур, которые подвергаются сдавлению. Вulbus – это название продолговатого мозга из-за его луковичной формы, поэтому бульбарный синдром означает признаки поражения продолговатого мозга.
А пирамиды – это анатомические образования продолговатого мозга, представляющие собой пучки нервных волокон, несущие импульсы от коры больших полушарий к нервным клеткам передних рогов спинного мозга. Пирамиды отвечают за произвольные движения в конечностях и туловище.
Соответственно вышеизложенному, бульбарно-пирамидный синдром клинически проявляет себя мышечной слабостью в конечностях, онемением и утратой болевой и температурной чувствительности (волокна проходят через продолговатый мозг).
Сдавление ядер черепно-мозговых нервов, располагающихся в стволе мозга, становится причиной возникновения расстройств зрения и слуха, речи (из-за нарушения движений языком), гнусавости голоса, поперхивания при принятии пищи, затруднения дыхания. Возможны кратковременные потери сознания или утраты мышечного тонуса при сохраненном сознании.
https://www.youtube.com/watch?v=pKPTPgn2MIk
Корешковый синдром в случае аномалии Арнольда-Киари заключается в появлении признаков нарушения функции черепно-мозговых нервов. Это могут быть нарушения подвижности языка, гнусавый или осиплый голос, нарушения проглатывания пищи, дефекты слуха (в том числе и шум в ушах), нарушения чувствительности на лице.
Синдром вертебробазилярной недостаточности связан с нарушением кровоснабжения в соответствующем кровеносном бассейне. Из-за этого возникают приступы головокружения, утраты сознания или мышечного тонуса, проблемы со зрением.
Как видим, становится ясно, что большинство симптомов аномалии Арнольда-Киари возникают не в результате одной непосредственной причины, а из-за сочетанного влияния различных факторов.
Так, приступы потери сознания обусловлены как сдавлением специфических центров продолговатого мозга, так и нарушением кровоснабжения в вертебробазилярном бассейне. Аналогичная ситуация возникает и с нарушением зрения, слуха, головокружением и так далее.
Сирингомиелитический синдром возникает не всегда, а только в случаях сочетания аномалии Арнольда-Киари с кистозными изменениями спинного мозга.
Эти ситуации проявляются диссоциированным нарушением чувствительности (когда изолированно нарушается температурная, болевая и тактильная чувствительность, а глубокая (положение конечности в пространстве) остается интактной), онемением и мышечной слабостью в некоторых конечностях, нарушениями функции тазовых органов (недержание мочи и кала). О том, чем проявляется сирингомиелия, Вы сможете прочитать в отдельной статье.
Каждая разновидность аномалии Арнольда-Киари имеет свои клинические особенности. Аномалия Арнольда-Киари I типа может никак себя не проявлять до 30-40 лет (пока организм молод, сдавление структур компенсируется). Иногда эта разновидность порока является случайной находкой при проведении магнитно-резонансной томографии по поводу другого заболевания.
II тип нередко сочетается с другими пороками: менингомиелоцеле поясничной области и стенозом водопровода мозга. Клинические проявления возникают с первых минут жизни. Помимо основных симптомов, у ребенка наблюдаются громкое дыхание с периодами его остановки, нарушение проглатывания молока, попадание еды в нос (ребенок давится, поперхивается и не может сосать грудь).
III тип также часто сочетается с другими пороками развития мозга и шейно-затылочной области. В мозговой грыже в шейно-затылочной области может располагаться не только мозжечок, но и продолговатый мозг, затылочные доли. Этот порок практически не совместим с жизнью.
IV тип некоторыми учеными, в последнее время, не считается симптомокомплексом Киари в современном представлении, потому что не сопровождается опущением недоразвитого мозжечка в большое затылочное отверстие. Однако классификация австрийца Киари, впервые описавшего эту патологию, содержит и IV тип.
Диагностика
Сочетание целого ряда симптомов, описанных выше, позволяет врачу заподозрить аномалию Арнольда-Киари.
Но для точного подтверждения диагноза необходимо проведения компьютерной или магнитно-резонансной томографии (последний метод информативнее).
Полученное с помощью магнитно-резонансной томографии изображение демонстрирует опущение структур задней черепной ямки ниже большого затылочного отверстия и подтверждает диагноз.
Лечение
Выбор метода лечения при аномалии Арнольда-Киари зависит от наличия симптомов заболевания.
Если порок был выявлен случайно (то есть не имеет клинических проявлений и не беспокоит больного) при проведении магнитно-резонансной томографии по поводу другого заболевания, то лечение не проводят вообще. За пациентом устанавливается динамическое наблюдение, чтобы не пропустить момент появления первых клинических симптомов сдавления мозга.
Если аномалия проявляет себя незначительно выраженным гипертензионно-гидроцефальным синдромом, то предпринимаются попытки консервативного лечения. Для этой цели используют:
- дегидратационные препараты (мочегонные). Они уменьшают количество ликвора, способствую уменьшению болевого синдрома;
- нестероидные противовоспалительные средства с целью уменьшения болевого синдрома;
- миорелаксанты при наличии напряжения мышц в шейной области.
Если применения лекарственных препаратов оказывается достаточно, то на какой-то период на этом и останавливаются.
Если же эффекта нет, или у больного появляются признаки других неврологических синдромов (мышечная слабость, утрата чувствительности, признаки нарушения функции черепно-мозговых нервов, периодические приступы потери сознания и так далее), то тогда прибегают к хирургическому лечению.
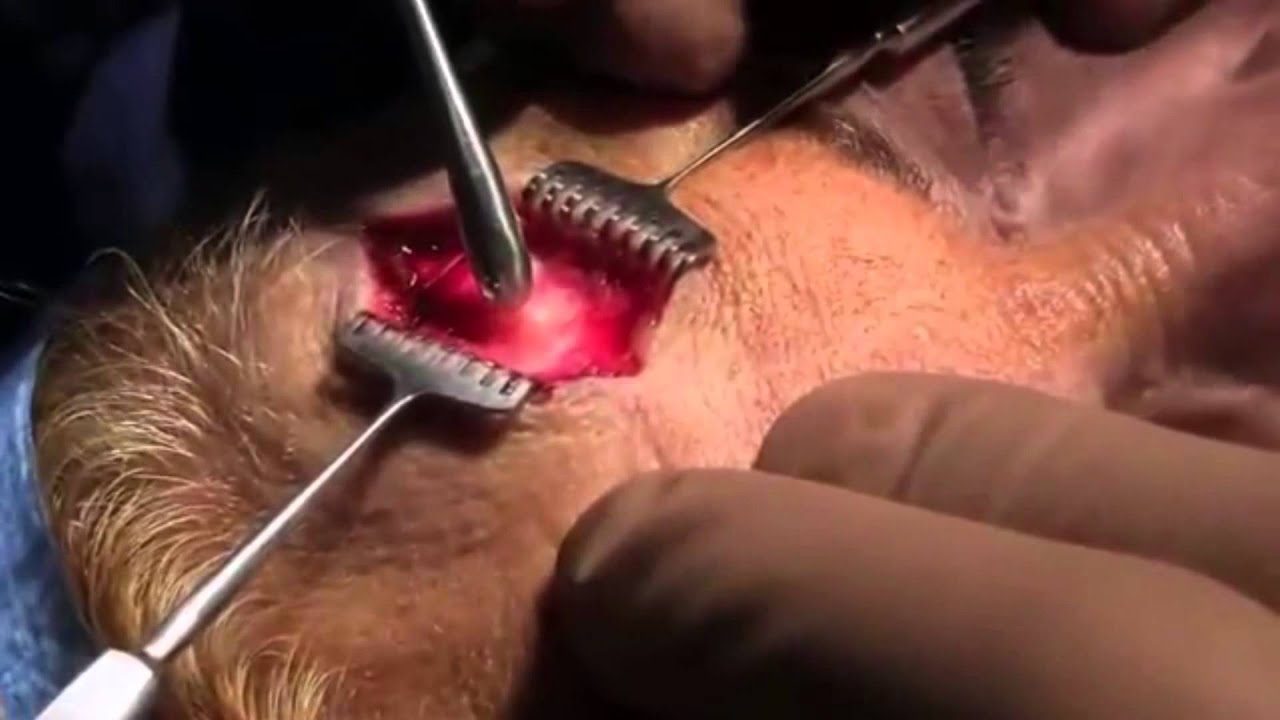
Оперативное лечение состоит в выполнении трепанации задней черепной ямки, удалении части затылочной кости, резекции опущенных в большое затылочное отверстие миндалин мозжечка, рассечении спаек субарахноидального пространства, мешающих циркуляции ликвора.
Иногда может понадобиться шунтирующая операция, целью которой является отведение избытка спинномозговой жидкости. «Лишняя жидкость» по специальной трубке (шунту) сбрасывается в грудную или брюшную полость.
Определение момента, когда возникает необходимость в хирургическом лечении, — весьма важная и ответственная задача. Длительно существующие изменения чувствительности, утрата мышечной силы, дефекты черепно-мозговых нервов могут не восстановиться полностью после оперативного вмешательства.
Поэтому важно не упустить момент, когда действительно без операции не обойтись. При II типе порока оперативное лечение показано практически в 100% случаев без предварительного консервативного лечения.
Таким образом, аномалия Арнольда-Киари – это один из пороков развития человека. Она может оказаться бессимптомной, а может проявить себя с первых дней жизни.
Клинические проявления заболевания весьма разнообразны, диагностика проводится с помощью магнитно-резонансной томографии. Лечебные подходы различны: от отсутствия какого-либо вмешательства до оперативных методов.
Объем лечебных мероприятий определяется индивидуально.
Источник: https://doctor-neurologist.ru/anomaliya-arnolda-kiari-simptomy-i-lechenie
Аномалия Арнольда-Киари
Аномалия Арнольда Киари – нарушение развития, которое заключается в виде несоразмерности размеров черепной ямки и структурных элементов мозга, располагающихся в ней. При этом мозжечковые миндалины спускаются ниже анатомического уровня и могут ущемляться.
Симптомы аномалии Арнольда Киари проявляются в виде частых головокружений, а иногда заканчиваются инсультом мозга. Признаки аномалии могут долго отсутствовать, а затем резко заявить о себе, например, после вирусной инфекции, удара головой или других провоцирующих факторов. Причем случиться это может на любом отрезке жизни.
Описание болезни
Сущность патологии сводится к неправильной локализации продолговатого мозга и мозжечка, в результате чего появляются краниоспинальные синдромы, которые врачи нередко расценивают как атипичный вариант сирингомиелии, рассеянного склероза, спинномозговой опухоли. У большинства больных аномалия развития ромбэнцефалона совмещается с другими нарушениями в спинном мозге – кистами, провоцирующими стремительную деструкцию спинномозговых структур.
Болезнь получила название в честь патологоанатома Арнольда Джулиуса (Германия), который описал аномальное отклонение в конце 18 века и врача из Австрии Ганса Киари, который изучал заболевание в тот же период времени. Распространенность нарушения варьируется в пределах 3–8 случаев на каждые 100000 человек. В основном встречается аномалия Арнольда Киари 1 и 2 степени, а взрослые с 3-м и 4-м типом аномалии живут совсем недолго.
Аномалия Арнольда Киари 1 типа заключается в опускании элементов задней черепной ямки в спинальный канал.
Болезнь Киари 2 типа характеризуется изменением местоположения продолговатого мозга и четвертого желудочка, при этом зачастую бывает водянка.
Гораздо реже встречается третья степень патологии, которой присущи выраженные смещения всех элементов черепной ямки. Четвертый тип представляет собой дисплазию мозжечка без его сдвига вниз.
Причины заболевания
По данным ряда авторов, болезнь Киари представляет собой недоразвитие мозжечка, сочетающееся с различными отклонениями в отделах мозга. Аномалия Арнольда Киари 1 степени – наиболее распространенная форма.
Это нарушение представляет собой одностороннее или двухстороннее опускание миндалин мозжечка в спинальный канал.
Это может произойти вследствие перемещения продолговатого мозга вниз, часто патология сопровождается различными нарушениями краниовертебральной границы.
Клинические проявления могут возникнуть только на 3–4 десятке жизни. При этом следует отметить, что бессимптомное течение эктопии миндалин мозжечка в лечении не нуждается и часто проявляется случайно на МРТ. На сегодняшний день этиология болезни, так же как и патогенез, изучены плохо. Определенная роль отводится генетическому фактору.
Выделяют три звена в механизме развития:
- генетически обусловленная врожденная остеоневропатия;
- травматизация ската во время родов;
- высокое давление ликвора на стенки спинномозгового канала.
Болезнь может быть вызвана наследственными нарушениями
Проявления
По частоте возникновения выделяют следующие симптомы:
- головные боли – у трети пациентов;
- боль в конечностях – 11%;
- слабость в руках и ногах (в одной или двух конечностях) – больше половины пациентов;
- чувство онемения в конечности – половина больных;
- снижение или утрата температурной и болевой восприимчивости – 40%;
- шаткость походки – 40%;
- непроизвольные колебания глаз – треть больных;
- двоение в глазах – 13%;
- нарушения глотания – 8%;
- рвота – у 5%;
- нарушения произношения – 4%;
- головокружения, глухота, онемение в лицевой области – у 3% больных;
- синкопальные (обморочные) состояния – 2%.
Боли в голове и области шеи – распространенный симптом патологии
Болезнь Киари второй степени (диагностируется у детей) сочетает в себе дислокацию мозжечка, ствола и четвертого желудочка. Неотъемлемый признак – наличие менингомиелоцеле в области поясницы (грыжа спинального канала с выпячиванием вещества спинного мозга).
Неврологическая симптоматика развивается на фоне аномального строения затылочной кости и шейного отдела позвоночного столба. Во всех случаях присутствует гидроцефалия, часто – сужение водопровода мозга. Неврологические признаки появляются с самого рождения.
Операция при менингомиелоцеле проводится в первые дни после рождения. Последующее хирургическое расширение задней черепной ямки позволяет добиться хороших результатов. Многие пациенты нуждаются в шунтировании, особенно при стенозе Сильвиевого водопровода.
При аномалии третьей степени черепно-мозговая грыжа внизу затылка или в верхней шейной области сочетается с нарушениями развития мозгового ствола, краниального основания и верхних позвонков шеи. Образование захватывает мозжечок и в 50% случаев – затылочную долю.
Эта патология встречается очень редко, имеет неблагоприятный прогноз и резко сокращает продолжительность жизни даже после операции.
Сколько именно человек будет жить после своевременного вмешательства, точно сказать нельзя, но, вероятнее всего, что недолго, так эта патология считается несовместимой с жизнью.
Четвертая степень заболевания представляет собой обособленную гипоплазию мозжечка и на сегодняшний день не относится к симптомокомплексам Арнольда-Киари.
Клинические проявления при первом типе прогрессируют медленно, в течение нескольких лет и сопровождаются включением в процесс верхнего шейного спинномозгового отдела и дистального отдела продолговатого мозга с нарушением работы мозжечка и каудальной группы черепных нервов. Таким образом, у лиц с аномалией Арнольда-Киари выделяют три неврологических синдрома:
- Бульбарный синдром сопровождается дисфункцией тройничного, лицевого, преддверно-улиткового, подъязычного и вагусного нервов. При этом наблюдаются нарушения глотания и речи, бьющий вниз спонтанный нистагм, головокружения, расстройства дыхания, парез мягкого неба с одной стороны, охриплость голоса, атаксии, дискоординация движений, неполный паралич нижних конечностей.
- Сирингомиелитический синдром проявляется атрофией мышц языка, нарушением глотания, отсутствием чувствительности в лицевой области, хриплостью голоса, нистагмом, слабостью в руках и ногах, спастическим повышением мышечного тонуса и т. д.
- Пирамидный синдром характеризуется незначительным спастическим парезом всех конечностей с гипотонусом рук и ног. Сухожильные рефлексы на конечностях повышаются, брюшные рефлексы не вызываются или снижаются.
Операция проводится при тяжелых формах нарушения
Боли в области затылка и шеи могут усиливаться при покашливании, чихании. В руках снижается температурная и болевая чувствительность, а также мышечная сила. Часто возникают обмороки, головокружения, у больных ухудшается зрение. При запущенной форме появляются апноэ (кратковременная остановка дыхания), быстрые неконтролируемые движения глаз, ухудшение глоточного рефлекса.
Интересный клинический признак у таких людей – провоцирование симптомов (синкопе, парестезии, боли и др.) натуживанием, смехом, кашлем, пробой Вальсальвы (усиленный выдох при закрытом носе и рте). При нарастании очаговых симптомов (стволовых, мозжечковых, спинномозговых) и гидроцефалии встает вопрос о хирургическом расширении задней черепной ямки (субокципитальной декомпрессии).
Диагноз аномалии первого типа не сопровождается повреждением спинного мозга и ставится в основном у взрослых посредством КТ и МРТ.
По данным патологоанатомического вскрытия, у детей с грыжей спинномозгового канала болезнь Киари второго типа выявляют в большинстве случаев (96–100%). С помощью УЗИ можно определить нарушения циркуляции ликвора.
В норме цереброспинальная жидкость легко циркулирует в подпаутинном пространстве.
Смещение миндалин вниз затрудняет циркуляцию. Из-за нарушения ликвородинамики возникает гидроцефалия.
Боковой рентген и МР картина черепа отображает расширение канала позвоночного столба на уровне С1 и С2. На ангиографии сонных артерий наблюдается огибание миндалины мозжечковой артерией. На рентгене отмечаются такие сопутствующие изменения краниовертебральной области, как недоразвитие атланта, зубовидного отростка эпистрофея, укорачивание атлантозатылочной дистанции.
При сирингомиелии на боковом снимке рентгена наблюдается недоразвитие задней дуги атланта, недоразвитие второго шейного позвонка, деформация большого затылочного отверстия, гипоплазия боковых частей атланта, расширение позвоночного канала на уровне С1-С2. Дополнительно следует провести МРТ и инвазивное рентгенологическое исследование.
МРТ – наиболее предпочтительный способ диагностики
Манифестация симптомов болезни у взрослых и лиц пожилого возраста часто становится поводом для выявления опухолей задней черепной ямки или краниоспинальной области.
В некоторых случаях правильно поставить диагноз помогают имеющиеся у пациентов внешние проявления: низкая линия оволосения, укороченная шея и др.
, а также наличие на рентгене, КТ и МРТ краниоспинальных признаков костных изменений.
Сегодня «золотым стандартом» диагностики нарушения является МРТ мозга и шейно-грудного отдела. Возможно внутриутробное проведение УЗИ диагностики. К вероятным ЭХО-признакам нарушения относятся внутренняя водянка, лимоноподобная форма головы и мозжечок в виде банана. В то же время некоторые специалисты не считают такие проявления специфичными.
Для уточнения диагноза используют различные плоскости сканирования, благодаря чему можно обнаружить несколько информативных в отношении болезни симптомов у плода. Получить изображение во время беременности достаточно легко. Ввиду этого УЗИ остается одним из основных вариантов сканирования для исключения патологии у плода во втором и третьем триместрах.
Выявление признаков заболевания у плода может стать показанием к исключению пороков развития позвоночного столба, однако даже абсолютное отсутствие данных УЗИ при этом указывает на расщепление позвоночника в 95% случаев.
При бессимптомном течении показано постоянное наблюдение с регулярным ультразвуковым и рентгенографическим исследованием.
Если единственный признак аномалии – незначительные боли, пациенту назначают консервативное лечение.
Оно включает разнообразные варианты с использованием нестероидных противовоспалительных средств и миорелаксантов. К наиболее распространенным НПВС относятся Ибупрофен и Диклофенак.
Нельзя самостоятельно назначать себе обезболивающие препараты, так как они имеют ряд противопоказаний (например, язвенная болезнь). При наличии какого-либо противопоказания врач подберет альтернативный вариант лечения. Время от времени назначают дегидратационную терапию.
Если в течение двух-трех месяцев эффекта от такого лечения нет, проводят операцию (расширение затылочного отверстия, удаление дужки позвонка и т. д.).
В этом случае требуется строго индивидуальный подход, позволяющий избежать как ненужного вмешательства, так и проволочки с операцией.
Тактика лечения в отношении каждого пациента требует индивидуального подхода
У некоторых пациентов хирургическая ревизия является способом постановки конечного диагноза. Цель вмешательства – ликвидация сдавливания нервных структур и нормализация ликвородинамики. Такое лечение приводит к существенному улучшению у двух-трех пациентов. Расширение черепной ямки способствует исчезновению головных болей, восстановлению осязаемости и подвижности.
Благоприятный прогностический признак – расположение мозжечка выше С1 позвонка и наличие только мозжечковой симптоматики. В течение трех лет после вмешательства могут возникать рецидивы. Таким пациентам по решению медико-социальной комиссии присваивается инвалидность.
Источник: https://spina.guru/bolezni/anomaliya-arnolda-kiari
Аномалия Киари
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие.
В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок.
Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии.
Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения.
Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие.
Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга.
Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения.
Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек.
Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи.
Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи.
Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом.
Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания.
Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос.
У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии.
Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию.
Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения.
Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки.
Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна.
Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов.
При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты.
Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия.
Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата.
В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу.
При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции.
По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источник: http://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/chiari-malformation
Аномалия Арнольда Киари 1 типа – что это? Симптомы, последствия, лечение
Не секрет, что во время формирования эмбриона в материнском лоне у него могут возникнуть разные патологии, в том числе и аномалия Арнольда Киари 1 типа.
Что это за болезнь? Медики называют это явление отклонением, которое развивается у плода из-за несоответствия размеров части черепной коробки и мозга, размещённого в ней. Речь идёт о зоне, где расположен мозжечок.
В результате происходит смещение в верхний отдел позвоночника части миндалин с дальнейшим их ущемлением.
Причины возникновения
Сможет ли нормально существовать человек, если у него обнаружена аномалия Арнольда Киари 1 типа? Продолжительность жизни, утверждают медики, зависит от нескольких факторов: степени тяжести и формы заболевания. Перед тем как ставить окончательный диагноз, они определяют причины, которые привели к развитию отклонения:
- Врождённые. Ещё в утробе у плода происходит деформация костей черепа: ямка, в которой находится мозжечок, оказывается слишком мала для растущего и развивающегося серого вещества. Среди других врождённых причин называют сильное увеличение затылочного отверстия у плода во время формирования скелета.
- Приобретённые. В первую очередь это травмы головы во время родов. Также к этой категории относят повреждения позвоночника: негативное влияние на него ликвора – особой жидкости, возникновение водянки и других патологий спинного мозга.
Если быть предельно откровенными, точные причины учёные пока не могут назвать. Единственное, в чём они уверены: синдром не связан с хромосомными аномалиями развития.
Факторы риска
Всем известно, что во время первого триместра беременности у плода идёт закладка фактически всех органов. Поэтому правильный образ жизни матери очень важен для нормального развития ребёнка. Если женщина не соблюдает элементарных правил, в дальнейшем это вызывает множество осложнений. Что касается синдрома Киари, то риск его возникновения увеличивают следующие факторы:
- Вредные привычки мамы: курение, злоупотребление алкоголем и наркотиками.
- Употребление сильнодействующих лекарств, самолечение.
- Перенесённые вирусные инфекции, особенно краснуха.
При особо лёгких формах болезни рождённый малыш не чувствует до конца жизни особых симптомов, особенно если у него диагностирована аномалия Арнольда Киари 1 типа.
Сколько живут дети, у которых третья или даже четвёртая стадия заболевания? Врачи дают неутешительные прогнозы. К сожалению, такие малыши в большинстве случаев обречены на летальный исход.
Даже если у младенца обнаружен первый или второй тип недуга, ему необходимо лечение: эффективность хирургического вмешательства составляет 85 %.
Типы синдрома
Независимо от своей тяжести, все они связаны с очень низким расположением мозжечка. Чем глубже и больше миндалины опускаются в позвоночный канал, тем более опасна и непредсказуема болезнь: вместе с ней диагностируют и другие отклонения. На основе этого и выделяют 4 формы патологии:
- Аномалия Арнольда Киари 1 типа – что это за разновидность болезни? Обычно она не вызывает какие-либо другие проблемы в развитии малыша. Структурных изменений в головном мозге не происходит, миндалины расположены в шейном отделе.
- Синдром второго типа. Мозжечок частично смещён в затылочное отверстие, при этом у эмбриона наблюдаются различные аномалии позвоночника, головного и спинного мозга.
- Заболевание третьего типа. Структуры заднего мозга полностью смещаются в затылочное отверстие. В этой зоне образуется грыжа.
- Заболевание Арнольда Киари четвертого типа. Для него характерна гипоплазия мозжечка – его недоразвитие. При этом он не смещается и часто сочетается с гидроцефалией и врождёнными кистами задней черепной ямы.
Медики говорят, что отклонения 2-го, 3-го и 4-го типа часто диагностируют в комбинации с другими серьёзными отклонениями в развитии малыша: гетеротопией коры мозга, гипоплазией подкорковых структур, аномалиями мозолистого тела и так далее.
Основные симптомы
Как уже говорилось, если стадия болезни очень лёгкая, а головной мозг задет незначительно, то болезнь может протекать фактически всю жизнь, не беспокоя человека. Но это достаточно редкий случай.
Обычно перманентные и регулярные головные боли – главный признак такого недуга, как аномалия Арнольда Киари 1 типа.
Симптомы для этой формы кроме мигрени такие: боль в шейном отделе, шум в ушах, тошнота и рвота, слабость рук, утрата чувствительности конечностей, мушки и двоение в глазах, неуверенная походка, смазанная речь и затруднённое дыхание.
Что касается синдрома второго типа, который является более опасным и сложным, то его характерные признаки проявляются сразу после рождения или в очень юном возрасте (часто в дошкольном).
Обычно это свистящее шумное дыхание у младенцев, слабый крик, нарушение дыхания и глотания. Таких новорождённых срочно реанимируют и пытаются спасти путём хирургического вмешательства.
В этих случаях главное – не упустить тот момент, когда ещё можно спасти жизнь ребёнку.
Другие признаки
Они наблюдаются при более тяжёлой степени синдрома. Первоисточниками инфаркта головного или спинного мозга часто бывают эти признаки аномалии Арнольда Киари: 1 тип не такой страшный, как его другие подвиды.
Для них характерно не только двоение в глазах, но и полная слепота, потеря сознания, нарушение координации, тремор конечностей, проблемы с мочеиспусканием, слабость мышц, потеря чувствительности большой части тела (иногда целой половины туловища).
Развитие разных последствий для здоровья может спровоцировать аномалия Арнольда Киари 1 типа. Продолжительность жизни и её нормальное течение зависят от быстрой и точной диагностики. Единственным методом обнаружить патологию является МРТ (магнитно-резонансная томография) мозга.
Она требует от пациента состояния полной неподвижности, поэтому активных младенцев усыпляют специальными медикаментами. При помощи этого аппарата также исследуют позвоночник, спинной мозг, шейные и грудные отделы скелета.
МРТ позволяет обнаружить не только синдром, но и все патологии, которые сопутствуют болезни.
Осложнения
Развитие различных патологий провоцирует аномалия Арнольда Киари 1 типа. Последствия могут быть самыми разными: начиная от хронического арахноидита и заканчивая паренхиматозным поражением аксона.
Особо ярко эти осложнения проявляются на фоне расстройства кровообращения в нервных тканях. Нейропсихические расстройства в этом случае проявляются поздно и проходят в форме отставания в развитии, параплегии.
Если аномалия развивается очень прогрессивно, она приводит к гидроцефалии – чрезмерному накоплению жидкости в желудочковой системе мозга.
Кроме того, синдром Арнольда Киари может вызвать такое сильное сдавливание спинного мозга, что человека полностью парализует.
Под его негативным воздействием часто начинают образовываться кисты и полости в позвоночнике: в них поступает жидкость, что нарушает работу спинного мозга. Также возможны осложнения с дыханием вплоть до полной его остановки.
Иногда медики регистрируют у пациентов застойную пневмонию – она становится последствием того, что человек теряет способность свободно двигаться.
Консервативное лечение
Синдром лечат двумя методами. Врачи выбирают консервативный или хирургический путь в зависимости от симптомов и признаков, проявляющихся у пациента, степени тяжести состояния человека, осложнений и последствий болезни.
Иногда исключительно при помощи медикаментов можно бороться с таким заболеванием, как аномалия Арнольда Киари 1 типа.
Что это за терапия? Во-первых, медик постоянно проводит профилактические осмотры больного, направляет его на консультации к разным специалистам, которые после соответствующего консилиума принимают необходимые меры.
Если пациент жалуется на боли в затылочной области головы или в шейном отделе, ему назначают обезболивающие и противовоспалительные средства, а также препараты, которые помогают расслабить мускулатуру.
Очень полезна и специальная лечебная физкультура – комплекс упражнений, направленный на устранение дрожи в конечностях и нормализацию координации. В этих целях также прописывают активную трудотерапию. А ещё будут полезны занятия с логопедом, который сумеет устранить проблемы с речью.
Все эти методы могут отсрочить или полностью предотвратить проведение более агрессивного лечения – операции.
Хирургическое вмешательство
Случается, что вышеназванные меры не помогают – активно продолжает развиваться и доставлять кучу хлопот аномалия Арнольда Киари 1 типа. Лечение тогда кардинально меняется: врач начинает готовить человека к оперативному вмешательству. Нейрохирургическая операция проводится с тремя целями.
Во-первых, для устранения таких серьёзных признаков болезни, как потеря сознания, расстройство зрения, слабость мускулатуры. Во-вторых, вмешательство помогает убрать первоисточник симптомов – ликвидировать сдавливание головного мозга. В-третьих, при помощи операции приводят в норму движение ликвора.
Вмешательство приостанавливает процесс изменений в структуре позвоночника и серого вещества: в результате симптомы ослабевают, пациент начинает чувствовать себя намного лучше.
Обычно хирурги из задней части черепной ямы удаляют фрагмент кости: пространство для мозга увеличивается, серое вещество перестаёт смещаться и сдавливаться. Также падает внутричерепное давление.
Кроме того, врачи активно используют такие методы, как шунтирование и закрытие позвоночного канала.
Профилактика
Чтобы у ребёнка не было вышеназванных проблем, будущая мама должна серьёзно относиться к своему положению. Во-первых, она обязана ознакомиться со всеми возможными патологиями, которые могут возникнуть у плода: нарушения развития нервной трубки, синдром Дауна, аномалия Арнольда Киари 1 типа.
Что это за болезни, почему они возникают и какие меры можно принять для уменьшения риска развития этих и других отклонений, должна знать каждая будущая мать. Грамотная и осведомлённая беременная женщина будет максимально избегать всех тех факторов, которые негативно влияют на формирование эмбриона.
Во-вторых, женщина обязана полноценно питаться, употреблять разнообразные полезные блюда: рыбу, мясо, фрукты, овощи, крупы и молочные продукты. Категорически не допускается алкоголь. Кроме того, нужно отказаться от курения, не говоря уже о наркотиках и сильнодействующих препаратах.
Любые лекарства, даже гомеопатические, будущая мама принимает только после назначения врача. В-третьих, женщина также должна много гулять, дышать свежим воздухом, много двигаться.
Про нервы, переживания и негативные эмоции стоит забыть, наслаждаясь своим положением и получая максимальную радость от жизни.
Источник: http://fb.ru/article/219694/anomaliya-arnolda-kiari-tipa---chto-eto-simptomyi-posledstviya-lechenie