- Синдром Апера
- Причины синдрома Апера
- Лечение синдрома Апера
- Прогноз и профилактика синдрома Апера
- Методы лечения синдрома Апера
- Причины возникновения синдрома Апера
- Характерные симптомы патологии
- Диагностика заболевания
- Лечение
- Возможные осложнения и прогноз
- Профилактические рекомендации
- Что такое синдром Апера и причины возникновения потологии
- Причины развития синдрома
- Симптоматика
- Диагностика
- Лечение синдрома Апера
- Вспомогательная терапия
- Синдром Апера
- Причины синдрома Апера
- Симптомы синдрома Апера
- Диагностика синдрома Апера
- Лечение синдрома Апера
- Прогноз и профилактика синдрома Апера
- Синдром Апера
- Причины синдрома Апера
- Симптомы синдрома Апера
- Диагностика синдрома Апера
- Лечение синдрома Апера
- Прогноз и профилактика синдрома Апера
Синдром Апера
Синдром Апера – генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей.
Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований.
Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания.
Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году.
Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных.
Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей.
Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной.
Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями.
Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R.
Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния.
Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки.
Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов.
Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR.
Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног.
Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь.
Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа.
Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие.
Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса.
Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты.
Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост.
При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов.
При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности.
Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию.
Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния.
Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни.
Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления.
По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект.
Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах.
Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется.
При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика синдрома Апера
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения.
При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов.
В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа.
Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком.
Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Источник:
http://www.krasotaimedicina.ru/diseases/genetic/Apert-syndrome
Методы лечения синдрома Апера
Синдром Апера представляет собой редкую генетическую патологию, выражающуюся в преждевременном внутриутробном закрытии костей черепа и дефектах костной системы. Клинически аномалия представлена необычной формой головы и лица, а также сращением пальцев на верхних (иногда и на нижних) конечностях. Патология встречается примерно у одного из 200 тысяч новорожденных.
Причины возникновения синдрома Апера
Симптомокомплекс, характерный для синдрома, впервые описал французский педиатр Эжен Апер (Аперт) в начале XX века. Аномалия также носит названия акрокраниодисфалангия, акроцефалосиндактилия, акросфеносиндактилия – все эти слова в переводе с латыни характеризуют отклонения от нормы в строении черепа, фаланг пальцев, описывая внешние признаки патологии.
Череп плода, находящегося в утробе матери, состоит из частей, которые ещё не полностью срослись между собой.
Между теменными и лобными костями находится коронарный (венечный) шов, а между теменными – стреловидный (сагиттальный).
Заращение этих соединительнотканных участков при описываемой аномалии происходит ранее, чем положено. Это приводит к появлению внешних признаков болезни и развитию внутренних патологий.
Мальчики с синдромом Апера рождаются также часто, как и девочки, то есть заболевание одинаково распространено среди представителей обоих полов. Основными факторами развития патологии считаются:
- Генная мутация. Изменение наследственных свойств в клетках организма одного из родителей в данном случае носит спонтанный характер. Ген, находящийся на десятой хромосоме и называемый FGFR2, расположен таким образом, что легко подвергается повреждениям. Именно этот участок ДНК отвечает за формирование и развитие соединительной (в частности, костной) ткани в эмбриональном периоде. Дефект гена приводит к заболеваниям костной системы. Вероятность рождения ребёнка с описываемым симптомокомплексом у пожилых родителей выше, чем у молодых.
- Наследственность. У человека, страдающего синдромом, с вероятностью 50% родится ребёнок с такой же аномалией.
- Рентгеновское облучение беременной женщины.
- Заболевания, перенесённые беременной: вирусы гриппа, туберкулёз, сифилис, краснуха и некоторые другие.
Синдром Апера, причины которого довольно сложно установить при отсутствии генетической предрасположенности, практически не поддаётся лечению.
Характерные симптомы патологии
Внешние признаки синдрома представляют чёткую клиническую картину, а в совокупности с другими проявлениями дают основание для постановки диагноза. Основные симптомы, определяемые у младенца в период новорожденности, вне зависимости от причин заболевания:
- Краниосиностоз – раннее сращение частей черепа – приводит к деформации черепных костей и определяет отклонение от нормы формы головы ребёнка. Такие дети имеют высокий выпуклый лоб, широкую переносицу, широко посаженные глаза, плоское лицо, выступающую нижнюю челюсть.
- Сращение пальцев на верхних и/или нижних конечностях перепончатое, костное либо кожное. Чаще всего соединёнными в процессе внутриутробного развития остаются указательный, средний и безымянный пальцы руки и аналогичные на ногах. Иногда три пальца срастаются настолько плотно, что на них формируется один общий ноготь.
- Дефекты внутриутробного развития костной ткани в ряде случаев приводят к карликовости.
- Позднее появление зубов, их деформация.
- Хроническая внутричерепная гипертензия, вызванная деформацией костей черепа, приводит к развитию слабоумия.
- У пациентов часто развиваются патологии внутренних органов: сердца, почек, поджелудочной железы. Нередко фиксируются аномалии развития позвонков, внешних половых органов. Страдает слух.
- Патологии органов зрения: косоглазие, вывих хрусталика, катаракта, нистагм, экзофтальм, пигментный ретинит и другие.
Совокупность клинических проявлений болезни позволяет сделать вывод о развитии синдрома Апера. На фото новорожденных, страдающих от патологии, видно сходство внешних черт.
Многие из описанных выше признаков разнятся в зависимости от условий жизни маленьких пациентов. Так, дети, от которых отказались родители, в детских учреждениях (детских домах, интернатах) показывают слабый уровень умственного развития. При этом ребёнок с диагнозом синдром Апера в возрасте 5 лет, живущий в семье, по уровню интеллекта может не уступать здоровым сверстникам.
Диагностика заболевания
Для постановки диагноза врачи изучают клиническую картину и данные анамнеза родителей. Из инструментальных методов применяют:
- Рентгенологические исследования. В младенческом возрасте показывают раннее закрытие швов черепа, во взрослом – деформацию черепных костей и позвоночника.
- Компьютерная томография черепа. Выявляет аномальное сращение костей (венечного шва), гипоплазию верхней челюсти, ненормальный размер глазниц и другие характерные признаки патологии.
- Магнитно-резонансная томография. Показывает степень изменения мягких тканей черепа.
- Ультразвуковые исследования плода. Некоторые отклонения внутриутробного развития (аномалии развития конечностей, костей черепа, пороки сердца) могут свидетельствовать о развитии синдрома Апера.
- Эхокардиография взрослых пациентов (УЗИ сердца) нередко показывает нарушения в работе сердечно-сосудистой системы: сердечную недостаточность, сужение просвета аорты и лёгочного ствола, дефект между правым и левым желудочками сердца.
- Молекулярно-генетический анализ ДНК. Высокоинформативный метод, позволяющий определить мутацию соответствующего гена.
Комплекс исследований позволяет провести дифференциальную диагностику с другими патологиями развития костной системы.
Корректная постановка диагноза имеет большое значение в связи с тем, что синдром Апера подразумевает лечение, отличное от терапии похожих заболеваний.
Лечение
Эффективного метода, который бы воздействовал на патогенез заболевания, не существует. Лечение носит паллиативный характер и направлено на минимизацию страданий пациента и улучшение качества жизни, насколько это возможно. Такую стратегию реализовывают посредством хирургического вмешательства и частично при помощи симптоматической медикаментозной терапии.
Операции при диагностированном синдроме выполняются по таким основным направлениям:
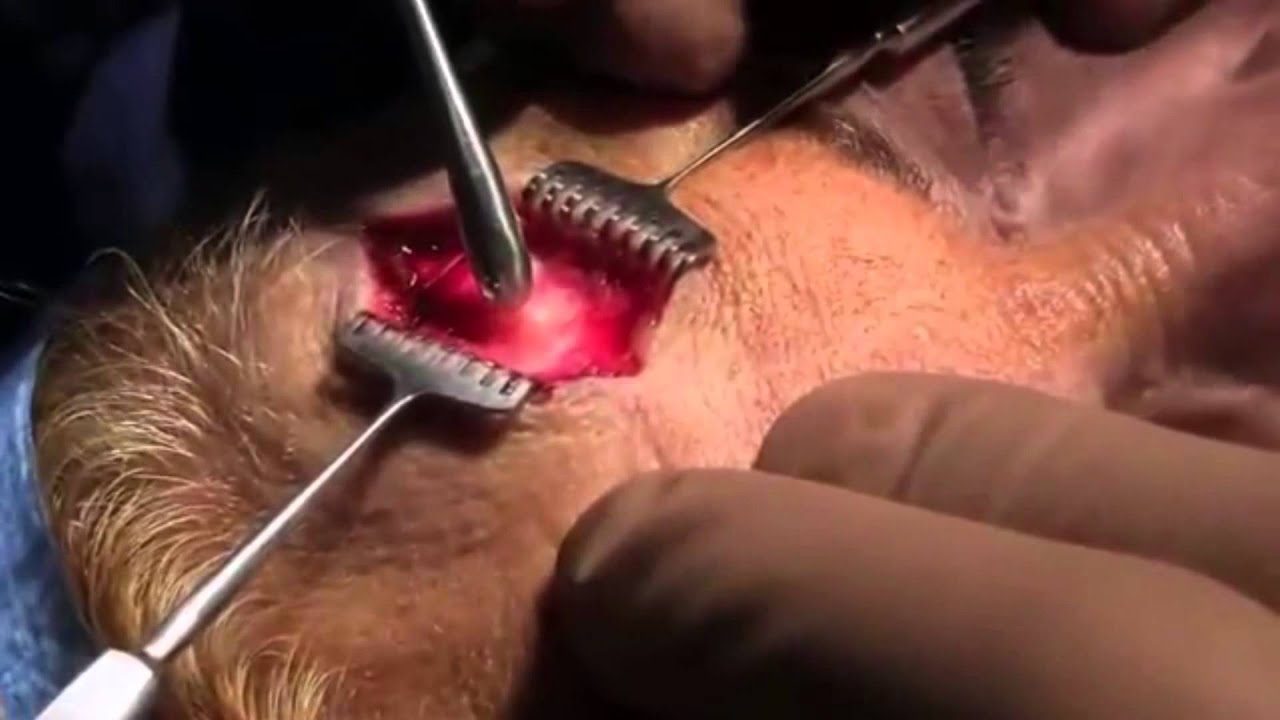
- Трепанация черепа. В процессе такой операции хирург производит разделение неправильно сросшихся частей черепа, восстанавливая его физиологическую форму. Хирургическое вмешательство иначе называется краниотомия, выполняется детям в возрасте около полугода. Оперативные манипуляции позволяют не только скорректировать форму головы, но и восстановить нормальное внутричерепное давление, а также избежать патологий, вызываемых гипертензией.
- Коррекция формы лица и глазного гипертелоризма. Операции преследуют преимущественно эстетические цели, проводятся в более позднем (от четырёх до двенадцати лет) возрасте.
- Хирургическое разделение пальцев. Производится в первые два года жизни ребёнка.
- Оперативное лечение сопутствующих симптомов (болезней сердца, органов зрения, слуха и так далее).
- Ортодонтические манипуляции с целью выравнивания прикуса и коррекции расположения и формы зубов.
Основную проблему – неправильно сросшиеся кости черепа, что провоцирует развитие черепной гипертензии – в современной медицине решают путём краниофациальной дистракции. Этот метод представляет собой постепенное растяжение костей черепа.
Медикаментозное лечение носит симптоматический характер. Пациентам назначают:
- Глазные капли и мази, снимающие патологическую сухость слизистых оболочек, а также лечащие сопутствующие патологии органов зрения.
- Препараты, снижающие внутричерепное давление.
- Антибиотики – против бактериальных инфекций верхних дыхательных путей, ушей, глаз, которым подвержены дети с синдромом Апера.
- Лекарственные средства, направленные на устранение сопутствующих симптомов, связанных с другими органами.
Эффективное лечение возможно лишь при условии слаженной работы врачей различных специализаций: хирурга, невролога, педиатра, офтальмолога, отоларинголога, психолога и других. Психическое здоровье пациента во многом определяется его адаптацией в социуме. Для решения этой проблемы привлекаются психотерапевты.
Возможные осложнения и прогноз
Вероятность развития осложнений на фоне отсутствия хирургического лечения высока. К наиболее вероятным из них относятся:
- развитие слабоумия;
- снижение зрения и слуха;
- заболевания позвоночника и суставов;
- патологии сердечно-сосудистой, выделительной и других систем.
Прогноз при отсутствии операции неблагоприятный, за исключением тех пациентов, которые дожили до средних лет, не имея пороков развития сердечно-сосудистой системы.
https://youtube.com/watch?v=Ak17q1p7XZ4
В случае своевременно проведённых оперативных вмешательств и лёгкой степени развития патологии пациент может жить до старости при условии, что мутация гена не повлекла за собой пороки сердца и тяжёлые патологии внутренних органов.
Профилактические рекомендации
Поскольку основной фактор развития синдрома – генные мутации, эффективной профилактики не существует. В данном контексте можно говорить о своевременной диагностике – УЗИ беременных женщин, особенно если в роду были случаи патологии.
Учитывая менее вероятные, но также существующие причины, к мерам профилактики можно отнести:
- недопущение рентгеновского облучения при беременности;
- своевременное лечение болезней при вынашивании плода.
Синдром Апера – редкая тяжёлая врождённая аномалия, не поддающаяся патогенетической терапии. Своевременное хирургическое и медикаментозное лечение носит паллиативный характер и может существенно улучшить качество жизни пациента.
(1 5,00 из 5)
Загрузка…
Источник:
https://ProSindrom.ru/genetic/sindrom-apera.html
Что такое синдром Апера и причины возникновения потологии
Синдром Апера или Аперта (Apert) представляет собой акроцефалосиндактилию и считается редко встречающейся патологией. Характерными проявлениями заболевания являются деформация костей черепа и конечностей, сопровождающаяся в половине случаев умственной отсталостью различной степени тяжести. В конце 19-го века синдром описал Уитон, а в начале 20-го века обобщен Апером.
Синдром Апера – генетическая аномалия, при которой швы черепных костей смыкаются раньше физиологического срока, что обуславливает изменение формы головы и лица. Заболевание имеет свойство передаваться из поколения к поколению по наследству.
Причины развития синдрома
Патология развивается в результате редчайшей генной мутации, приводящей к нарушению формирования костных структур и их фиксации в период внутриутробного развития. Наряду с генетикой, основанием появления синдрома считают облучение будущей матери рентгеновскими лучами или перенесенные в течении беременности инфекции (туберкулез, менингит, краснуха, грипп).
Чаще всего патология встречается у детей возрастных родителей. При этом вероятность его наследования превышает 50%.
Симптоматика
Большинство клинических проявлений становятся заметны при рождении младенца. Это связано с тем, что развиваются они в период пребывания плода в материнской утробе. К основным признакам патологии относятся:
- Деформация черепных костей («башенный» череп, вытянутый вверх, широко расставленные и слегка выпученные глаза, неправильный прикус с сильно выступающей верхней челюстью, широкий нос);
- Сращивание пальцевых фаланг на конечностях – чаще остальных срастаются I, II, III, пальцы посредством формирования между ними кожной перепонки или, нередко, истинного сращивания;
- На кистях, стопах присутствуют дополнительные пальцы;
- Умственная отсталость, задержка психического развития, отмечающиеся у 100% пациентов;
- Атрофия зрительного нерва, иногда провоцирующая абсолютную слепоту;
- Стабильная внутричерепная гипертензия возникает в силу преждевременного сведения швов черепных костей. Сопровождается тошнотой, головокружением, рвотой;
- На фоне недоразвитой верхней челюсти развивается нарушение дыхания;
- Эмоциональная нестабильность (несдержанность, импульсивность, агрессия).
Диагностика
На приеме врач, исходя из жалоб и анамнестических данных, выясняет, встречалась ли данная аномалия у членов семьи.
Диагностические мероприятия включают в себя:
- Неврологический осмотр, при котором врач оценивает форму черепа и интеллектуальные способности пациента (если это возможно) посредством беседы и заполнения специально разработанных анкет;
- Осмотр глазного дна. Рассеянность дисковых контуров зрительного нерва и его отечность указывают на высокое внутричерепное давление;
- Рентгенография костей черепа, позволяющая оценить состояние и форму черепных костей;
- Рентгенография конечностей (стоп и кистей) определяет причину сращивания кожной перепонки между фалангами или костей. Данное исследование имеет важную роль на этапе планирования хирургического вмешательства;
- Магнитно-резонансная (МРТ) и компьютерная (КТ) томографии головы дают возможность послойно оценить состояние черепных костей и головного мозга, а также выявить симптомы преждевременного сращения черепных швов и даже гидроцефалию (избыточное скопление цереброспинальной жидкости, за счет которой происходит питание мозговых клеток) вследствие повышенного внутричерепного давления.
В некоторых случаях пациенту необходимо проконсультироваться у медицинского генетика и нейрохирурга.
Лечение синдрома Апера
В настоящий момент радикальной терапии синдрома Аперта не существует. Симптоматическая терапия основывается на проведении краниотомии – вмешательства, позволяющего изменить форму черепных костей и в необходимых пределах увеличить объем черепа.
Оперативное лечение предусматривает следующую последовательность хирургических вмешательств:
- Ремоделирующая краниотомия, при которой хирург отделяет сросшиеся костные фрагменты и выполняет частичную репозицию. Подобное вмешательство допустимо к осуществлению в период достижения маленьким пациентом возраста 6 – 9 месяцев. Благодаря краниопластике полностью восстанавливается подвижность черепных костей, обеспечивающая возможность дальнейшего роста головного мозга, минуя возникновение внутричерепной гипертензии.
- Лицевая пластика, при которой хирургом корректируется форма верхней челюсти и скуловых костей, формируется правильная форма лица. Данный вид хирургического лечения доступен к выполнению у детей от 5 до 14 лет. В случае проведения первичной пластики в раннем возрасте, маленькому пациенту может потребоваться дополнительное корректирующее вмешательство. Пластическая операция лицевого отдела способствует не только эстетической коррекции, но стабилизации процесса дыхания, который иногда может быть нарушен на фоне деформированной верхней челюсти.
- Коррекция глазного гипертелоризма, подразумевающая сокращение расстояния между глазными яблоками из-за расширенного корня носа. Параллельно врачами может быть произведена дополнительная пластика нижней челюсти с целью изменения ее формы, и выдвижение верхней челюсти.
Вспомогательная терапия
Вспомогательные мероприятия при синдроме Апера включают в себя следующие манипуляции:
- Закапывание глазных капель на протяжение дня (способствует устранению опасного пересыхания глазного дна), а также закладывание мази на время ночного сна.
- СИПАП-терапия – регулярное использование аппарата, способствующего поддержанию стабильного положительного давления в дыхательных путях. Ребенку с синдромом Апера, страдающему обструктивным апноэ ночного сна, рекомендовано использование маски, которая подключена к специальной аппаратуре. Данный прибор способен создать положительное давление на все протяжении дыхательных путей, гарантируя нормализацию просвета бронхов в период сна.
- Антибактериальная терапия. Дети, страдающие синдромом Аперта, подвержены частому возникновению бактериальных инфекций носа и ушей, лечение которых предусматривает назначение антибактериальных средств.
- Трахеостомия (фиксация в трахее дыхательной трубки). Вмешательство показано детям, больным синдромом Аперта, состояние которых осложнено тяжелой формой обструктивного апноэ сна.
- Миринготомия (внедрение трубки в барабанную перепонку). Применяется в качестве дополнительного вмешательства у детей с затянувшимися или часто повторяющимися инфекциями ушей бактериального характера.
Источник:
https://sustavi.guru/sindrom-apera.html
Синдром Апера
Синдром Апера – генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей.
Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований.
Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания.
Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году.
Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных.
Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей.
Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной.
Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями.
Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R.
Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния.
Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки.
Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов.
Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR.
Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног.
Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь.
Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа.
Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие.
Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса.
Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты.
Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост.
При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика синдрома Апера
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов.
При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности.
Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию.
Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния.
Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни.
Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления.
По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект.
Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах.
Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется.
При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика синдрома Апера
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения.
При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов.
В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа.
Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком.
Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Источник:
Синдром Апера
Синдром Апера – генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей.
Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований.
Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания.
Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году.
Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных.
Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей.
Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной.
Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями.
Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R.
Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния.
Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки.
Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов.
Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR.
Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног.
Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь.
Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа.
Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие.
Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса.
Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты.
Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост.
При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика синдрома Апера
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов.
При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности.
Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию.
Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния.
Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни.
Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления.
По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект.
Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах.
Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется.
При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика синдрома Апера
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения.
При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов.
В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа.
Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком.
Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Источник: